使用人类iPS细胞科学家们确定了导致ALS的某些患者的遗传变异
名古屋大学的研究人员在一些肌萎缩侧索硬化症(ALS) 患者中发现了一种新的基因变异。他们使用人类诱导的多能干细胞 (iPS),详细描述了这种变异与肌萎缩侧索硬化相关的过程。研究人员期望这种机制成为 ALS 治疗的新治疗靶点。研究结果发表在《神经科学杂志》上。
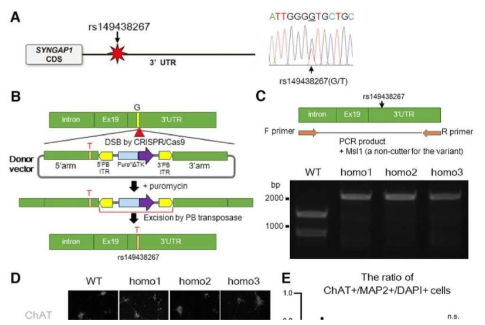
ALS 是一种进行性和致命的神经退行性疾病,患者会逐渐失去运动神经元并出现肌肉无力。这种疾病的机制和原因尚不清楚,也没有治疗方法。此外,ALS 是一种多样化的疾病;不同的患者似乎有不同的原因、机制和症状。研究人员知道,一种叫做融合肉瘤 (FUS) 的蛋白质在这种疾病中起着关键作用。通常,FUS 与 RNA 结合并调节 RNA 的功能。另一方面,FUS 功能障碍与各种神经退行性疾病有关,包括 ALS。
名古屋大学医学研究生院神经病学系的横井聪博士及其同事一直在研究这种 FUS 蛋白。此前,他们发现 FUS 蛋白与编码一种名为 SYNGAP1 的蛋白质的 RNA 相互作用。SYNGAP1 有助于突触形成,这对于神经元协同工作至关重要。“目前,没有研究报道 SYNGAP1 参与 ALS 的机制。但是,鉴于它与 FUS 的密切关系,我们想调查 SYNGAP1 是否与 ALS 有任何关系,”该论文的第一作者 Yokoi 博士说。学习。
首先,研究小组在 ALS 患者中寻找 SYNGAP1 基因的变体。他们发现 807 名患者中有 7 名携带该变异体。接下来,为了检查 SYNGAP1 变体的行为,他们在人类 iPS 衍生的运动神经元中复制了这个变体基因。
iPS 细胞是一种干细胞,科学家可以将其转化为体内任何类型的细胞,包括神经元。它在医学研究中特别有用,因为研究人员可以生成例如患病细胞,并在活体状态下对它们进行不同的测试。
这与研究人员在动物细胞或活生物体外部进行测试的传统方法形成对比。
在这项研究中,研究人员从 iPS 细胞中生成了带有 SYNGAP1 变体的运动神经元。在这些神经元中,研究人员观察到与正常运动神经元相比的几种异常行为。特别是,该变体过度募集了 FUS 蛋白,以及另一种称为 HNRNPK 的 RNA 结合蛋白。这种过度结合抑制了突触形成。
特别是,HNRNPK 而非 FUS 的过度募集似乎是 SYNGAP1 变体突触功能障碍的主要原因。此外,当研究人员应用反义 RNA 阻断 HNRNPK 与 SYNGAP1 RNA 的过度结合时,他们恢复了突触形成,这意味着运动神经元可以协同工作。这表明,在未来,科学家们可以利用反义 RNA 来开发 ALS 药物。
HNRNPK 与 SYNGAP1 的过度结合导致突触异常,这解释了发生突触丢失时早期 ALS 的过程。此外,虽然以前的 ALS 研究主要集中在 FUS,但本研究强调了 SYNGAP1 和 HNRNPK 的关键作用。目前研究中使用的反义RNA只对那些带有变异SYNGAP1的患者有效。尽管如此,研究人员希望这种新机制的发现能够为其他类型的 ALS 提供新的见解。
有趣的是,这项研究还发现,与用于 SYNGAP1 研究的小鼠模型相比,SYNGAP1 在人类 iPS 细胞中的表现不同。“我们认为使用人源样本至关重要,这样来自这些细胞的观察结果可以直接应用于患者,”横井博士说。
尽管研究人员描述了肌萎缩侧索硬化的一种机制,但由于肌萎缩侧索硬化涉及多种不同的机制和原因,因此仍有许多问题有待研究。“我在日常实践中看到的 ALS 患者尚未接受治愈性治疗。我们将继续研究以找到可以应用于未来 ALS 治疗的方法,”Yokoi 博士说。
版权声明:本文由用户上传,如有侵权请联系删除!