人工智能有助于探索化学前沿
在原子水平上模拟系统行为的能力代表了从药物设计到材料发现等各个领域的强大工具。由洛斯阿拉莫斯国家实验室研究人员领导的团队开发了机器学习原子间势,可以预测作用于原子的分子能量和力,从而实现与现有计算方法相比节省时间和费用的模拟。
洛斯阿拉莫斯化学物理学家、最近一篇描述这项工作的《自然化学》论文的合著者本杰明·内布根 (Benjamin Nebgen) 表示:“机器学习的潜力越来越多地为计算成本高昂的模拟提供了一种有效的替代方案,这些模拟试图在原子尺度上表示复杂的物理系统。” “通用反应机器学习原子间势,适用于广泛的反应化学,无需改装,将极大地有利于化学和材料科学。”
缩小有效模拟的差距
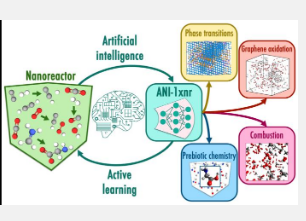
传统上,为化学中的分子动力学建立有效的模拟是通过基于物理的计算模型来完成的,包括经典力场或量子力学。虽然量子力学模型是准确且普遍适用的,但它们的计算成本极其昂贵。相比之下,经典力场计算效率高,但精度相对较低,并且仅适用于有限范围的系统。 ANI-1xnr 是该团队的变革性机器学习模型,弥补了化学领域数十年来在速度、准确性和通用性方面的差距。 (机器学习是人工智能的一种应用,计算机程序通过训练“学习”。)
ANI-1xnr 代表了第一个足够通用的反应式机器学习原子间势 - 它可以应用于许多不同的化学系统 - 可以与基于物理的计算模型竞争以执行大规模反应原子模拟。 ANI-1xnr 是使用自动化工作流程开发的,该工作流程对包含碳、氢、氮和氧元素的各种化学系统进行反应分子动力学模拟。
事实证明,ANI-1xnr 能够研究各种系统,从碳相变到燃烧,再到生命起源前的化学。该团队通过将模拟结果与实验和传统计算技术的结果进行比较来验证模拟结果。
转化原子间势
“ANI-1xnr 不需要领域专业知识或针对每个新用例进行改装,使来自不同领域的科学家能够研究未知的化学,”洛斯阿拉莫斯计算科学家、该论文的共同通讯作者 Richard Messerly 说。 “ANI-1xnr 的普遍适用性是变革性的,代表着朝着取代大规模研究反应化学的长期建模技术迈出了重要一步。”
该团队使用的数据集和 ANI-1xnr 代码已向研究界公开。
免责声明:本文由用户上传,与本网站立场无关。财经信息仅供读者参考,并不构成投资建议。投资者据此操作,风险自担。 如有侵权请联系删除!
-
2025年6月20日,——在世界文化遗产地河南洛阳的光影流转之间,2025年新浪微博旅游之夜盛大举行。作为国内首个...浏览全文>>
-
2025年6月20日,——在世界文化遗产地河南洛阳的光影流转之间,2025年新浪微博旅游之夜盛大举行。作为国内首个...浏览全文>>
-
QQ多米试驾线下预约活动为了让更多用户感受QQ多米的独特魅力,我们特别推出了线下试驾预约活动。这不仅是一次...浏览全文>>
-
阜阳长安启源A07以其卓越的性能和豪华配置吸引了众多消费者的目光。作为一款定位高端市场的新能源车型,长安启...浏览全文>>
-
【安徽淮南大众CC新车报价2025款大公开】大众CC作为一款兼具运动感与豪华质感的轿跑车型,一直深受消费者喜爱...浏览全文>>
-
2025款长安猎手K50在安徽淮南地区的最新价格已新鲜出炉,为准备购车的朋友带来全面解析。这款车型以其高性价比...浏览全文>>
-
在安徽滁州购买长安猎手K50时,了解其落地价和省钱技巧至关重要。长安猎手K50是一款实用性强的皮卡车型,适合...浏览全文>>
-
途锐新能源是大众旗下的一款高端插电混动SUV,目前在安徽阜阳地区有售。其官方指导价约为58万元起,但实际成交...浏览全文>>
-
2025款大众CC作为一款兼具运动与豪华的中型轿车,备受关注。目前市场指导价大约在25万至35万元之间,具体价格...浏览全文>>
-
2024款探岳X作为一款备受关注的中型SUV,在市场上以其时尚的设计和出色的性能吸引了众多消费者。根据最新市场...浏览全文>>
- QQ多米试驾线下预约
- 安徽滁州长安猎手K50落地价,买车省钱秘籍
- 淮南大众CC新款价格2025款多少钱?买车攻略一网打尽
- 瑞虎8 PRO试驾,畅享豪华驾乘,体验卓越性能
- 安徽阜阳长安启源A05多少钱 2025款落地价,换代前的购车良机,不容错过
- 保时捷Macan试驾的流程是什么
- 安徽淮南大众ID.3多少钱?购车攻略在此
- 阜阳揽巡落地价,豪华配置超值价来袭
- 安徽池州威然 2024新款价格与配置的完美平衡
- 奇瑞瑞虎9试驾,新手必知的详细步骤
- QQ多米价格,换代前的购车良机,不容错过
- 池州迈腾GTE新款价格2022款多少钱?选车秘籍与优惠全公开
- 岚图追光多少钱 2024款落地价走势,近一个月最低售价25.28万起,性价比凸显
- 天津滨海威然 2024新款价格,最低售价28.98万起,入手正当时
- 蚌埠途昂新款价格2025款多少钱?购车必看
- 坦克400预约试驾全攻略
- 天津滨海ID.7 VIZZION价格,各配置车型售价全揭晓,性价比之王
- 安庆帕萨特最新价格2025款,最低售价12.35万起,入手正当时
- 亳州宝来新款价格2025款多少钱?选车指南与落地价全解析
- 生活家PHEV 2025新款价格,最低售价63.98万起现在该入手吗?