宏基因组测序超快速GPU加速病原体识别方法
宏基因组测序 (mNGS) 是一种强大的诊断工具,可在临床微生物检测中检测致病病原体。宏基因组序列的快速准确分类是 mNGS 测试干实验室步骤中病原体鉴定的关键程序。但是,可以通过在临床相关时间范围内对序列进行分类来改进这一关键步骤。
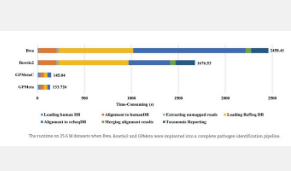
为了应对这一挑战,王学斌领导的华大基因团队最近推出了 GPMeta,这是一种超快速病原体检测方法,并在生物信息学简报中发表了这些亮点。
GPMeta可以通过复杂海量的mNGS测序数据,快速准确地识别病原体。使用来自临床样本的模拟数据集和宏基因组测序数据集,将结果与生物信息学研究社区使用的工具(例如 Bowtie2、Bwa、Kraken2 和 Centrifuge)进行了基准测试。
结果表明,GPMeta 不仅具有更高的精度,而且速度也明显更快。此外,GPMeta 提供了 GPMetaC 聚类算法,这是一种用于聚类和重新评分模糊比对的统计模型,以提高对平均核苷酸同一性 >95% 的微生物基因组中高度同源序列的区分。这些结果强调了GPMeta 在开发需要快速周转时间的传染病 mNGS 测试中的关键作用。
背景
更快、更早地检测致病病原体对于精确的抗生素治疗而不是经验性治疗至关重要。它可以在一次测试中同时检测出患者体内几乎所有新的和已知的病原微生物,在感染诊断方面具有巨大的应用潜力。
mNGS 检测包括两部分:湿实验室实验操作,涉及临床样本预处理、总核酸提取、文库制备和测序,以及干实验室生物信息学分析,包括原始测序数据预处理、去除人类宿主序列、序列比对到精选数据微生物序列的病原体数据库和分类学分类。
生物信息学分析是mNGS检测最后的关键步骤,需要快速、准确地完成,以加速整个检测过程。然而,迫切需要新的策略来加速病原体鉴定的生物信息学分析。
为应对这一挑战,GPMeta采用了简洁的哈希索引方案,支持多GPU同时对分库进行拆分,满足了人们日益增长的处理快速膨胀的微生物基因组能力的需求。
免责声明:本文由用户上传,与本网站立场无关。财经信息仅供读者参考,并不构成投资建议。投资者据此操作,风险自担。 如有侵权请联系删除!