人类泛基因组参考将使对基因组多样性的理解更加完整和公平
加州大学圣克鲁兹分校的科学家与一组研究人员一起发布了第一个人类泛基因组的草稿——这是一个新的、可用的基因组学参考,它结合了来自不同祖先背景的 47 个人的遗传物质,以便更深入、更准确地理解全球基因组多样性。
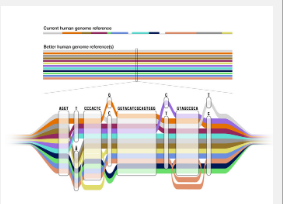
通过将 1.19 亿个碱基(DNA 序列中的“字母”)添加到现有的基因组学参考中,泛基因组提供了人类遗传多样性的表示,而这在单个参考基因组中是不可能实现的。正如今天发表在《自然》、《基因组研究》、《自然生物技术》和《自然方法》杂志上的一系列开创性论文所示,它非常准确、更完整并显着增加了对人类基因组变异的检测。
泛基因组由人类泛基因组参考联盟 (HPRC) 制作,该联盟由 UCSC 生物分子工程副教授 Benedict Paten 和生物分子工程助理教授 Karen Miga 共同领导,现在可用于 UCSC 基因组的组装中心浏览器。十几名 UCSC 研究人员和学生是该项目的贡献者,该项目将持续到 2024 年,届时研究人员计划发布包含 350 个人基因组信息的最终泛基因组。
“我们通过对不同的人进行抽样并将他们纳入这种每个人都可以使用的结构中,从而在参考文献中引入更多的多样性和公平性,”主要标记论文的资深作者帕滕说。“一个基因组不足以代表所有人——泛基因组最终将成为具有包容性和代表性的东西。”
了解基因组变异
每个人的基因组略有不同——与下一个人相比平均相差约 0.4%——了解这些差异可以深入了解他们的健康状况,有助于诊断疾病、预测医疗结果和指导治疗。使用泛基因组参考将提高科学家在未来研究中检测和理解变异的能力。
通常,当科学家和临床医生研究个体的基因组以寻找变异时,他们会将个体的 DNA 与标准参考的 DNA 进行比较,以确定一个或多个碱基对的差异所在。到目前为止,参考基因组主要由每个人类染色体的单个序列表示,主要来自一个个体。但是,这个参考已经有将近 20 年的历史了,而且从根本上来说是有限的,因为它不能代表人类中存在的丰富的遗传变异。这在基因组分析中引入了一个称为参考偏差的问题。
相比之下,新的泛基因组是一个参考,它结合了来自不同祖先背景的 47 个个体的基因组。泛基因组在序列具有相同碱基的区域看起来像线性参考,并扩展以显示存在差异的区域。它同时代表了人类基因组序列的许多不同版本,并为科学家提供了一个更准确的比较点,用于比较某些人群中存在的变异,而不是其他人群中存在的变异。
“一个基因组不可能代表我们知道可以在世界各地观察和研究的所有丰富变异,”UCSC HPRC 生产中心主任 Miga 说。“人类泛基因组参考文献的首要目标是尝试扩大参考资源的代表性,使其更包容、更公平地研究人类物种,作为参考文献的集合,而不仅仅是一个。”
基因组变异可以很小,仅由一个或几个 DNA 碱基的差异组成,也可以是较大的结构变异,归类为 50 个碱基对或更大的变异。这些较大的结构变异可能对健康产生重要影响。到目前为止,由于技术有限和使用单一参考序列的偏见,研究人员无法识别人类基因组中超过 70% 的结构变异。
在泛基因组参考中添加的 1.19 亿个新碱基中,大约有 9000 万个来自结构变异。结构变异很复杂,可能是序列倒置、插入、缺失或串联重复——两个或多个碱基重复多次的片段。这些新碱基将帮助研究人员研究基因组中以前没有参考的区域,并有可能在未来的研究中将结构变异与疾病联系起来。
“现在,我们可以映射到更多的结构变异,因此我们正在基因组中寻找以前不存在的特征和区域,”Miga 说。“这很令人兴奋,因为它让我们能够以一种我们以前无法研究的独特方式来研究基因调控,因为这些区域可能会被不恰当地绘制或完全忽略。”
与使用标准参考的检测相比,使用泛基因组参考进行基因组分析可将结构变异的检测提高 104%。由于泛基因组中存在的数据量增加,泛基因组参考还提高了识别小变体(那些只有几个碱基长)的准确性约 34%。
每个人都携带一对染色体——一组遗传自母亲,一组遗传自父亲。泛基因组参考中的个体基因组包含单倍型解析信息,这意味着它可以自信地区分两组父母的染色体——这是一项重大的科学壮举。掌握这些信息将有助于科学家更好地了解各种基因和疾病是如何遗传的。
免责声明:本文由用户上传,与本网站立场无关。财经信息仅供读者参考,并不构成投资建议。投资者据此操作,风险自担。 如有侵权请联系删除!